
What is Coloboma.

Before talking about coloboma renal, let’s get to know what is coloboma. Coloboma is derive from the Greek terminology “Koloboma, meaning mutilated, i.e., something that is severely damage”. Hence, coloboma renal is a sporadic genetic condition that primarily interferes with an individual’s kidney (renal coloboma syndrome) and eye development (ocular coloboma); however, it exerts its influence on other organs as well, including the central nervous system, spine, limbs, gastrointestinal tract, skin etc. As the primary target of coloboma is the renal system and eyes, this article is centrally incline towards this syndrome.
Renal Coloboma Syndrome.
Renal coloboma syndrome, which is scientifically termed papillorenal syndrome, is a genetic condition that affects the development of kidneys at the fetal stage. This will result in small and undeveloped kidneys called hypoplasia kidneys. It directly leads to another condition called ESRD (end-stage renal disease). ESRD is mark by inappropriate filtration of waste through kidneys, and in 10% of cases, the root cause of ESRD is renal coloboma syndrome or papillorenal syndrome.
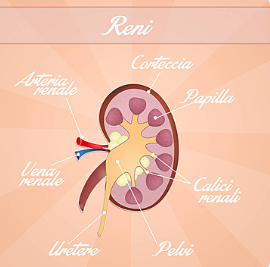
Here, it would help if you kept in mind that most people with such syndrome have visual malfunctions as well due to developmental issues in the optic nerve. Now, although this is distinctly known as ocular coloboma due to its simultaneous detection with papillorenal coloboma, it is often term as renal-eye disease.
Renal Coloboma Syndrome Prognosis.
Wondering what happens to the kidneys if you have renal coloboma. The frequently evident features of this disease involves malformation, which may lead to insufficient kidney function due to abnormally formed and small kidneys, known as renal hypoplasia. Tissue culture studies predict that in the coloboma renal, kidneys may have oligomeganephronia (i.e., considerably enlarged glomeruli).
Other renal outcomes include a horseshoe kidney (a condition where the lower end of the kidneys is fuse together) and a multicystic dysplastic kidney. Further consequences of coloboma renal may include proteinuria, hypertension, and vesicoureteral reflux.
Moreover, an expected prolonged renal insufficiency may lead to end-stage renal disease, which can cause severely dysplastic kidneys prenatally. In such cases, oligohydramnios may lead to fetal loss during pregnancy.
kidney failure can happen at any stage of life, leading to dialysis.
Renal Coloboma Causes.
The papillorenal syndrome has a genetic basis, so it is mainly cause by a mutation in a gene, the PAX-2 gene. PAX-2 gene primarily encodes proteins that are responsible for healthy development of eyes, ears, brain and kidneys. This underdevelopment leads to the malfunctioning of respective organs. However, the fact that the kidneys and eyes are majorly affected by the mutation is still unclear.
Renal Coloboma Syndrome Gene Review.
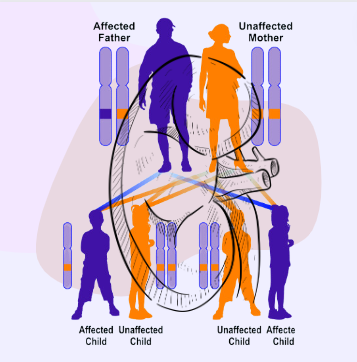
Now you know that coloboma renal is a genetic disorder. This syndrome is inherited in an autosomal dominant fashion. By autosomal, we mean the concerned gene is located on non-sex chromosomes, so babies of both genders can be affected by the disease. However, by dominant, we suggest that only one gene from the allele pair is enough to cause the disease, which can clearly reflect the critical nature of this syndrome.
Molecular testing reveals a vast range of phenotypes linked with pathogenic variants of the culprit gene PAX-2. Such variants are likely link with both ocular and renal abnormalities but are increasingly recognizes in cases which relates to coloboma renal.
The recurrent findings linked to PAX-2 gene-related disorders involve abnormalities of kidney structure and function. Other includes:
Hypoplastic kidneys. Diagnosed by ultrasound and characterized by one or both kidneys smaller in size.
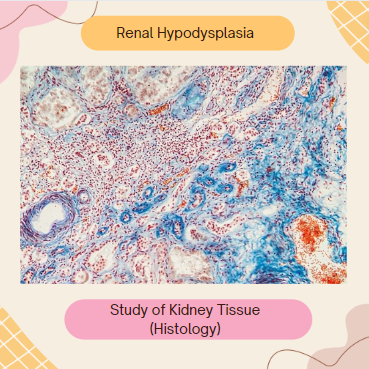
Renal hypodysplasia. Analyzing histologically involves a smaller kidney size with disorganized renal tissues and fewer nephrons.
Multicystic dysplastic kidney. Observes histologically in almost 6% of individuals encounters damage PAX-2 gene and is characterize by dysplastic or disorganize kidneys.
Other renal anomalies are congenital anomalies of the kidney and urinary tract (CAKUT). They usually involve medullary sponge kidney, horseshoe kidney, renal malrotation, and ureteropelvic junction obstruction.
Uncommon non-CAKUT. It usually involves nephrolithiasis (Kidney stones) and rarely an intrarenal teratoma.
Symptoms of Renal Coloboma Syndrome.
Some commonly detected signs of coloboma renal include the following.
- Visual impairment or ocular coloboma (it must be clear that this symptom is appear in maximum patients but not necessarily in all)
- Renal abnormalities, including impairment.
- Smaller glomeruli or oligomeganephronia.
- Hypertension.
- Development of multiple cysts in kidneys.
- Gradual hearing loss.
- Loose joints.
- Kidney failure at later stages.
- Fetal loss in case of oligohydramnios.
- Vesicoureteral reflux (when urine goes back from the bladder to the kidneys causes increase amount of ionophores in kidneys)
Treatment.
Coloboma renal has a genetic basis, so no specific therapy is suggestive to cure the condition. However, the emphasis is laid on managing the symptoms of syndrome. In this regard, hypertension management via medication and kidney failure management via dialysis is centrally focused during coloboma treatment. Additionally, a long-term visit to a nephrologist for the management of any future complication is highly suggestive.
Coloboma-associated Syndromes.
There are a number of conditions which result from coloboma, such as:
- Charge syndrome. A constellation of coloboma associated with growth retardation, heart defects, ear and genital abnormalities and microphthalmia (small eye).
- Manitoba oculotrichoanal syndrome. It involves multiple anomalies associated with anal opening, hair and ear.
- Fraser syndrome. Characterized by webbing of fingers and toes along with genital and renal abnormalities.
- Goldenhar syndrome. It involves faulty development of the nose, jaw, ear and soft palate.
- Amniotic band syndrome. The tangling of the fibrous band of the amniotic sac around the growing fetus.
- Cat-eye syndrome. Well known as Schmid-Fraccaro syndrome, it is characterized by kidney malformation, coloboma eye, heart defects, mild intellectual disability, and dysmorphic facial features.